
 There is no cure for AAT deficiency, but behavioral and lifestyle modifications, coupled with drug therapy and AAT replacement therapy and the promise of gene-replacement therapy, offer hope
There is no cure for AAT deficiency, but behavioral and lifestyle modifications, coupled with drug therapy and AAT replacement therapy and the promise of gene-replacement therapy, offer hope
Alpha1-antitrypsin (AAT) is a serum protein that is produced in the liver. It functions to protect the lungs so that they can work normally. The destruction of elastin that occurs in chronic obstructive pulmonary disease (COPD) is believed to result from an imbalance between two proteins in the lung: an enzyme called neutrophil elastase, which breaks down elastin, and AAT, which inhibits elastase.1 In healthy people, there is usually enough AAT to protect elastin, so abnormal elastin destruction does not occur. When, however, there is a genetic deficiency of AAT, the activity of the elastase is not inhibited, and elastin breakdown occurs.
AAT deficiency, first described in 1964 by Laurell and Eriksson,2 is a hereditary disorder characterized by decreased levels of this plasma protein. People with AAT deficiency have an increased risk of early-onset COPD, which can appear in those as young as 30 to 40 years old (particularly if they smoke).3
AAT deficiency is an autosomal codominantly inherited disorder occurring in about 1 in every 3,000 births.4 Carrying the trait does not invariably lead to the disease. Each parent must contribute one abnormal gene to produce offspring with AAT deficiency. It is estimated that only 1% to 3% of all cases of COPD are due to AAT deficiency.5
Pathogenesis
The AAT protein is encoded by a gene (PI) located on chromosome 14.6 This plasma glycoprotein is secreted primarily by liver cells and, to a lesser extent, by bronchial epithelial cells and macrophages. In healthy individuals, the function of AAT is to inhibit a variety of proteases, including elastase secreted by alveolar neutrophils. The neutrophils enter the lung and release elastase in response to infection or inflammation, as part of the innate immune response. In the alveoli, AAT binds elastase to its active site. Once elastase is bound, AAT alters its conformation and, in so doing, traps and permanently inactivates elastase. A deficiency of plasma AAT results in unrestrained protease digestion of the alveolar walls. The elastase also stimulates the alveolar-macrophage release of leukotriene B4, resulting in further neutrophil recruitment and amplified tissue destruction; this, in turn, results in the clinical manifestations of chronic lung disease as early as the third decade of life, especially in smokers.7,8
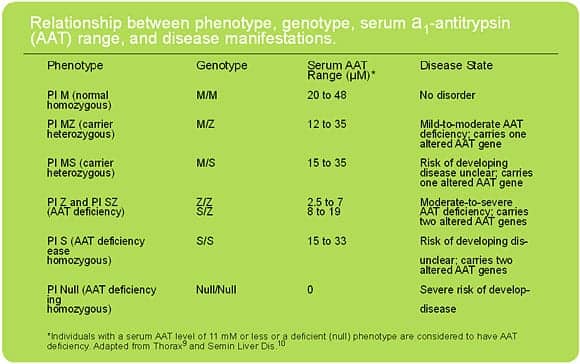
Genetic Transmission
Genetic transmission of AAT deficiency follows a Mendelian pattern of co-dominant inheritance. Homozygous and heterozygous conditions occur, and individuals who have one normal and one abnormal AAT gene are carriers. They have AAT levels that might be lower than normal but not as low as the deficiency state, and their risks of significant health problems are much lower than the risks of those with severe deficiency (see Table).9,10
The most common variants, occurring with a frequency of 95% in the United States,11 are the normal M-allele variants. The S variants are most frequent among individuals of Spanish or Portuguese descent, and the Z variants are most common in patients of Northern or Western European descent.12 In the United States, AAT deficiency is one of the three most common lethal genetic diseases among white adults11; cystic fibrosis and Down syndrome are the other two.11 Severe AAT deficiency affects an estimated 100,000 individuals in the United States, and approximately 25 million people carry at least one defective gene.13 Internationally, AAT deficiency has been identified in all populations but is more common in individuals of Northern European and Iberian descent.14 Similar rates are found among whites worldwide, with an estimated 117 million carriers and 3.4 million affected individuals.13,14
Identification of Individuals at Risk
A combination of environmental and genetic factors influences the susceptibility of an AAT-deficient individual to developing COPD. The most important environmental risk factor for COPD is cigarette smoking, which increases neutrophil elastase production, but individuals vary in their vulnerability to the effects of cigarette smoke. Severe AAT deficiency is a proven genetic risk factor for COPD.15 AAT-deficient phenotypes, however, are present in only 1% to 2% of COPD patients.15 This observation implies that COPD probably represents a complex genetic disease with multiple genetic and environmental contributions. Individuals with severe AAT deficiency vary in the likelihood of developing COPD and in the severity of COPD, which suggests that genetic and environmental interactions may be relevant to disease expression.
Early symptoms of AAT deficiency are generally respiratory in nature. Initial symptoms include cough, sputum production, and wheezing. Symptoms are initially intermittent and, if wheezing is the major symptom, individuals are often told they have asthma. The symptom that eventually dominates AAT deficiency is dyspnea. As in other forms of emphysema, the dyspnea of AAT deficiency is initially evident only with strenuous exercise. Over the years, however, it may progress, and eventually limit even mild activities. If COPD is seen in nonsmokers of any age, or if COPD occurs earlier than expected in smokers, then the probability of AAT deficiency as the underlying cause is increased.16
Chest radiographs of individuals with AAT-deficiency emphysema typically show the lungs as hyperlucent because healthy tissue has been destroyed. This process is not uniform; certain areas are affected more than others. The affected lung regions also might be described as oligemic because they lack the normal pattern of branching blood vessels.17 In some PI Z patients, the emphysema has a striking basilar distribution; in contrast, cigarette smoking is associated with more severe apical disease.18
High-resolution CT scanning may be used in the diagnosis of emphysema, but it typically does not distinguish between the conventional and AAT-deficient forms of COPD. In cases of moderate-to-severe COPD, high-resolution CT typically shows hypo-attenuating areas resulting from the destruction of lung architecture.
A laboratory blood test can determine whether a person has low serum levels of the protective AAT protein, and can help the clinician diagnose the cause of early-onset COPD (especially when a patient does not have obvious risk factors such as smoking or exposure to lung irritants). AAT phenotype testing can be done as a follow-up to determine the amount and type of AAT being produced. In addition, a DNA-based cheek swab test has been developed recently, and can be used when an AAT abnormality is established.18 The genetic testing determines which PI alleles are present. The test does not screen for every variant, just the most common ones (M, S, and Z, as well as any that are common for a particular geographical area or family). Once the affected individual’s PI-gene alleles have been determined, other family members should be tested to determine their own possible risk of developing emphysema and/or liver involvement, as well as the likelihood that their children might inherit the mutation.
 Treatment
Treatment
There is no cure for AAT deficiency at this time. There are, however, several forms of treatment, including behavioral and lifestyle modification, drug therapy for lung problems, and specialized therapy for AAT deficiency.
Smoking cessation is the first priority in managing patients with AAT deficiency. Lifelong nonsmokers could have a good chance of avoiding serious lung disease, even with AAT deficiency; and current smokers should stop smoking upon diagnosis.
Avoiding environmental pollutants is also advisable. Any home, occupational, or environmental airborne particulates smaller than 10 µm can cause further lung irritation and worsen the current condition of patients with AAT deficiency.19
Development of an exercise and nutrition program will increase stamina, will improve mental outlook, and might help to preserve lung and liver function.20 Annual immunizations for influenza and vaccination against pneumonia are important for protection of the vulnerable lungs.
Aggressive treatment of lung infections also is recommended, due to the increased neutrophil elastase release during microbial insult.17 Antibiotics can help to speed recovery, and avoidance of people who are sick and children under 5 years of age is recommended to prevent initial infections.17
Bronchodilators are often useful for maximizing lung function, and inhaled corticosteroids can help control symptoms. Corticosteroids enter cells and bind to steroid receptors that modulate transcription controlling protein expression.21 In addition, they reduce the release of inflammatory mediators and interfere with neutrophil activity.21
Oxygen supplementation also can be useful in patients with moderate-to-severe AAT-deficiency COPD. Reducing hypoxemia may decrease mortality rates in patients with severe airflow obstruction, and oxygen supplementation can prolong survival for this group.22
In 1987, Wewers et al23 showed that intravenous infusion of pooled human plasma AAT at a dose of 60 mg per kg, administered once weekly, produced serum AAT levels that generally exceeded the protective threshold. That same year, replacement therapy (also called augmentation therapy) was approved.24 Currently, three purified human a1-proteinase inhibitor preparations are available for replacement therapy. Long-term treatment with any of these preparations can improve pulmonary function, but it is unclear whether such treatment can retard the progression of AAT-deficiency COPD.25-27
AAT replacement therapy must be taken throughout a patient’s life. The therapy is expensive (costing approximately $52,000 per year) and inconvenient, with weekly injections required.28 Because of the lack of definitive data showing improvement in survival, there is some resistance on the part of third-party payors to cover the cost of treatment. Existing cost-effectiveness data are derived from observational, registry-based studies.28
Replacement therapy is intended only for AAT-deficient patients who have begun to show symptoms of COPD. It is not recommended for people without AAT deficiency who develop COPD as a result of cigarette smoking or other environmental factors.
Treatments aimed at correcting the underlying genetic abnormality, supplementing or modifying the gene product, and halting or reversing lung and liver injury are now beginning to emerge. In animal studies,29,30 investigators have successfully transferred a normal human AAT gene into striated muscle cells using an adeno-associated virus vector. Other investigations are aimed at turning off production of the endogenous abnormal gene product. For example, in one study,31 researchers tried to shut down the production of variant AAT proteins by using antisense oligonucleotides and ribozyme technology to interfere with translation of the mRNA message for the mutant protein. While results with cell cultures and animal studies are promising, the effectiveness of these approaches in humans is still unknown. Speculative approaches to treatment include gene repair (correcting targeted single-site gene mutations using RNA/DNA probe technology) and a stem-cell-based approach to AAT deficiency. Both of these innovative approaches are in their infancy. Therapeutic success with any one of these innovative approaches might one day prove effective, however, at eliminating both lung and liver disease associated with AAT deficiency.
Conclusion
AAT deficiency is a well-recognized cause of pulmonary problems, including early-onset COPD. Currently, the cornerstones of treatment are smoking cessation, conventional COPD treatment, and AAT replacement therapy. Therapeutic modalities currently under investigation are directed toward correcting the underlying genetic abnormality, supplementing or modifying the gene product, and halting or reversing pulmonary destruction.
Phyllis C. Braun, PhD, is a contributing writer for RT. For more information, contact [email protected].
References
1. Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. 2005; 365:2225-2236.
2. Laurell CB, Eriksson S. Hypo-alpha1-antitrypsinemia. Verh Dtsch Ges Inn Med. 1964; 70: 537-539.
3. Abboud RT, Ford GT, Chapman KR. Emphysema in alpha1-antitrypsin deficiency: does replacement therapy affect outcome? Treat Respir Med. 2005; 4:1-8.
4. Silverman EK, Chapman HA, Drazen JM, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med. 1998; 157:1770-1778.
5. Eden E, Mitchell D, Mehlman B, et al. Atopy, asthma, and emphysema in patients with severe alpha1-antitrypysin deficiency. Am J Respir Crit Care Med. 1997; 156:68-74.
6. Seixas S, Garcia O, Trovoada MJ, Santos MT, Amorim A, Rocha J. Patterns of haplotype diversity within the serpin gene cluster at 14q32.1: insights into the natural history of the alpha1-antitrypsin polymorphism. Hum Genet. 2001; 108:20.
7. Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004;170:1172-1178.
8. Hill AT, Bayley DL, Campbell EJ, Hill SL, Stockley RA. Airways inflammation in chronic bronchitis: the effects of smoking and alpha1-antitrypsin deficiency. Eur Respir J. 2000; 15:886-890.
9. Mahadeva R, Lomas DA. Genetics and respiratory disease. 2. Alpha1-antitrypsin deficiency, cirrhosis and emphysema. Thorax. 1998; 53:501-505.
10. Sifers RN, Finegold MJ, Woo SL. Molecular biology and genetics of alpha1-antitrypsin deficiency. Semin Liver Dis. 1992; 12:301-310.
11. Jardi R, Rodriguez-Frias F, Lopez-Talavera JC, et al. Characterization of the new alpha1-antitrypsin-deficient PI M-type allele, PI M (Vall d’Hebron) (pro(369)-ser). Hum Hered. 2000; 50:320-321.
12. Shahid A, Siddiqui AA, Zuberi SJ, Wagar MA. Genetic variants of serum alpha1-antitrypsin. J Pak Med Assoc. 1995; 45:245-246.
13. Miravitlles M. Alpha1-antitrypsin deficiency: epidemiology and prevalence. Respir Med. 2000; 94(suppl): S12-S15.
14. Blanco I, de Serras FJ, Fernandez-Bustillo E, Lara B, Miravitlles M. Estimated numbers and prevalence of PI*S and PI*Z alleles of alpha1-antitrypsin deficiency in European countries. Eur Respir J. 2006; 27:77-84.
15. DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2. Genetic aspects of alpha1-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004; 59:259-264.
16. Piitulainen E, Sveger T. Respiratory symptoms and lung function in young adults with severe alpha1-antitrypsin deficiency (PiZZ). Thorax. 2002; 57:705-708.
17. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2003; 168: 818-900.
18. Teckman JH, Lindblad D. Alpha1-antitrypsin deficiency: diagnosis, pathophysiology, and management. Curr Gastroenterol Rep. 2006; 8:14-20.
19. Koenig JQ. Pulmonary reaction to environmental pollutants. J Allergy Clin Immunol. 1987; 79:833-843.
20. Dowson LJ, Newall C, Guest PJ, Hill SL, Stockley RA. Exercise capacity predicts health status in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2001; 163:936-941.
21. MacDonald JL, Johnson CE. Pathophysiology and treatment of alpha1-antitrypsin deficiency. American Journal of Health-System Pharmacy. 1995; 52: 481-489.
22. Stoller JK, Tomashefski J Jr, Crystal RG, et al. Mortality in individuals with severe deficiency of alpha1-antitrypsin: findings from the National Heart, Lung, and Blood Institute registry. Chest. 2005; 127:1196-1204.
23. Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987; 316: 1055-1062.
24. American Lung Association. Alpha1-related emphysema. 2005. Available at: www.lungusa.org/site/apps/s/content.asp?c=dvLUK9O0E&b=34706&ct=3004003. Accessed September 25, 2006.
25. Abusriwil H, Stockley RA. Alpha1-antitrypsin replacement therapy: current status. Curr Opin Pulm Med. 2006; 12:125-131.
26. Abboud RT, Ford GT, Chapman KR. Emphysema in alpha1-antitrypsin deficiency: does replacement therapy affect outcome? Treat Respir Med. 2005; 4:1-8.
27. Ad Hoc Committee on Alpha1-Antitrypsin Replacement Therapy, Standards Committee, Canadian Thoracic Society. Current status of alpha1-antitrypsin replacement therapy: recommendations for the management of patients with severe hereditary deficiency. Can Med Assoc J. 1992; 146:841-844.
28. Alkins SA, O’Malley P. Should health-care systems pay for replacement therapy in patients with alpha1-antitrypsin deficiency? A critical review and cost-effectiveness analysis. Chest. 2000; 117:875-880.
29. Song S, Scott-Jorgensen M, Wang J, et al. Intramuscular administration of recombinant adeno-associated virus 2 alpha1-antitrypsin (rAAV-SERPINA1) vectors in a nonhuman primate model: safety and immunologic aspects. Mol Ther. 2002; 6:329-335.
30. Song S, Morgan M, Ellis T, et al. Sustained secretion of human alpha1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. Proc Natl Acad Sci U S A. 1998; 95:14384-14388.
31. Sandhaus RA. Alpha1-antitrypsin deficiency: 6. New and emerging treatments for alpha1-antitrypsin deficiency. Thorax. 2004; 59:904-909.






