 |
Interstitial lung disease (ILD), an umbrella diagnosis also referred to as diffuse parenchymal lung disease, encompasses a plethora of parenchymal lung disorders, such as farmer’s lung, hot tub lung, humidifier lung, sarcoidosis, black lung pneumoconiosis, and the very little understood idiopathic interstitial pneumonias (IIP), including idiopathic fibrosis. Diseases considered “interstitial” are lumped together based on similarities in clinical presentation, radiographic features, and physiology. The vast array of causes associated with ILDs makes diagnosis and identification of its etiology very difficult, and at times impossible. The intent of this synopsis is to present the classification scheme of ILDs, common pathophysiology, and the diagnostic tests available to the clinician for identifying them.
Classification of ILDs
Interstitial lung disease affects between 31.5 and 26.1 per 100,000 American men and women, respectively.1 The most commonly diagnosed type of ILD is idiopathic pulmonary fibrosis or IPF—responsible for 25% to 35% of all ILD cases.2 It is further estimated that 75% of patients diagnosed with ILD are further diagnosed as having IPF, sarcoidosis, or connective tissue disease.2 Of those ILDs with known etiology, the most common are drug related, environmental inhalants, and connective tissue diseases.2 The Table is a summary of causes for ILD categorized as known or unknown etiology. Represented in each of the known causes are a multitude of sources in which a specific agent may be identified at the time ILD is diagnosed. The IIPs listed were classified as idiopathic by a panel consensus of both the European Respiratory Society (ERS) and the American Thoracic Society (ATS). This classification scheme for IIPs by the ERS and ATS lists specific high resolution computed tomography (HRCT) findings associated with histological patterns. This classification scheme is considered the “gold standard” for diagnosis of IIPs.4
| Known Etiology | Unknown Etiology |
| Inorganic dusts Drugs Toxins Oxygen Radiation exposure Infections Hypersensitivity reaction |
Idiopathic interstitial pneumonia2 Idiopathic pulmonary fibrosis Nonspecific interstitial pneumonia Cryptogenic organizing pneumonia Respiratory bronchiolitis ILD Acute interstitial pneumonia Lymphoid interstitial pneumonia Collagen vascular disease Sarcoidosis |
The search for a specific etiologic agent of ILD requires a detailed history and physical examination, along with diagnostic testing such as a HRCT and/or surgical lung biopsy. Following a thorough evaluation to rule out all known causes of ILD with no definitive cause found, a diagnosis of IIP can be made.5 Idiopathic interstitial pneumonias include idiopathic pulmonary fibrosis, cryptogenic organizing pneumonia, respiratory bronchiolitis ILD, nonspecific interstitial pneumonia (NSIP), desquamative interstitial pneumonia, lymphocytic interstitial pneumonia, and acute interstitial pneumonia.2
Although a consensus has been reached on classification, one author suggests classification that is based on cellular versus fibrotic processes.5 This classification highlights the pathology of some IIPs along with other causes of ILD, such as cellular drug reactions and cellular pneumonitis, as “cellular” processes not resulting in fibrosis. This scheme lays the foundation for good to intermediate to poor prognosis depending on the degree of cellularity versus fibrosis associated with the specific ILD. This classification may not isolate IPF from the other IIPs if another IIP existed that had the extensive fibrosis seen in IPF. This can be seen in collagen vascular disease and drug reactions that resemble IPF on HRCT.5 The ERS/ATS classification scheme does present specific HRCT findings, listing whether fibrosis is present in linear reticulations and without architectural distortion or fibrosis with distortion as in IPF or the absence of fibrosis.
The diagnosis of ILD is made simple when the causal agent is known and can be isolated, helping to minimize lung damage and in some instances allow for healing. The diagnosis of the idiopathic interstitial lung pneumonias is difficult, although a solid classification such as that agreed upon by members of the European Respiratory Society and the American Thoracic Society has been laid out by two outstanding medical communities.
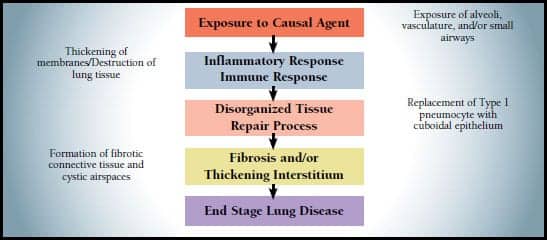 |
| Figure. Pattern of disease development. |
Unifying Pathological Process
The various causes of ILD, whether organic dusts, drugs, infections, or sarcoidosis, all follow identical patterns of disease development—a process ending ultimately with the development of a thickened interstitium that may or may not have fibrotic changes (see Figure). The first step in the development of such an aggressive disease is an exposure or predisposition to a causal agent. This initial exposure induces lung injury such as that seen in adult respiratory distress syndrome (ARDS) and its development from exposure to high levels of oxygen. Injury to the lung then induces an immediate and often aggressive immune response releasing cytokines and other inflammatory mediators, resulting in inflammation of parenchymal tissue responsible for gas exchange. The body then responds with a repair process that results in a thickened interstitium, fibrosis, or cystic airspaces.
Diagnostics for ILD
Diagnosis of an ILD, as mentioned earlier, requires an extensive investigation into the patient’s symptoms, with life style, work history, exposures, and medications forming the clinical context. The majority of patients will present with a respiratory symptom of cough or dyspnea on exertion. Dyspnea on exertion rather than at rest is most associated with a parenchymal disorder of the lung rather than a cardiopulmonary disorder. One author suggests summarizing gathered data for diagnosis and evaluating it using three pivotal parameters: 1) clinical context; 2) tempo of the disease process; and 3) radiographic findings.2 This framework does not include a surgical lung biopsy, which might give a more definitive etiology of the disease. When a specific diagnosis can be made within the three parameters, a lung biopsy may not be required. If, however, a diagnosis cannot be made by utilizing the three parameters, a lung biopsy must be considered.2
Even though the above framework has been presented, surgical lung biopsy is still considered by some to be the gold standard for the diagnosis of parenchymal lung disease.6 However, it is important to note that conclusiveness of the surgical biopsy is based on sample size, site selection, and expertise of the pathologist.7 Computed tomography scans should be used to determine the site from which the biopsy should be collected.7 A specimen should include an area where affected tissue borders less affected tissue. This allows the clinician to see the pathological process frozen in time—comparing damaged tissue with less damaged tissue.7 One study8 in Iceland with 73 patients concluded:
- The complication rate of surgical lung biopsy was 16%;
- 30-day operative mortality was 2.7%;
- Both mortality and complications increased in patients with preoperative respiratory failure; and
- The clinical diagnosis was changed in 73% of patients and the course of treatment was changed in 53% of patients following surgical lung biopsy.
Therefore, surgical lung biopsy is not without risk, and the benefits must be weighed. There is great debate over the need for surgical biopsies in all patients with clinical and radiographic indications for the diagnosis of ILD. The risk is increased in patients who have already encountered respiratory failure. A second study with 200 patients indicated a postprocedural mortality rate of 4.3%, and those with acute exacerbation during the biopsy experienced a 28.6% mortality rate. The study concluded that risk factors for mortality included patients with acute exacerbations and a lower DLco on pulmonary function tests.9
For lung biopsies, not only would variability lie in the quality and quantity of the specimen, but also in the interobserver of that specimen. One study found that the diagnosis between local pathologists and expert pathologists differed by 52%. This resulted in changes made to disease management in 60% of the cases.10 One source indicates that there is histological variability in surgical lung biopsies, stating that often the diagnoses of NSIP and IPF are made in multiple lobes and in some cases the same lobe.10 What are the chances that both IIPs would exist in the same lobe? This represents the limitations of pathologists in the diagnosis of specific IIPs. There remain two other options for the collection of tissue samples: video assisted thoracoscopy (VATS) and bronchoscopy. Patients who are not mechanically ventilated and immunocompromised experienced a mortality rate as low as 1.5% with VATS.10
Bronchoscopy has not been proven to be a reliable and effective procedure for the diagnosis of IIPs. There are limitations to the size of the biopsy that can be obtained and its ability to provide a sample large enough to identify interstitial pneumonias.7 Bronchoscopy with alveolar lavage and transbronchial biopsy does provide the ability to rule out sarcoidosis, hypersensitive pneumonitis, and any infection-related cause.7 Therefore, it may be prudent and beneficial to perform a biopsy to rule out these potential diagnoses. The ERS and ATS diagnostic pathway does include bronchoscopy prior to open-lung biopsy.
Advancement in the area of radiology and more specifically with CT has given great aid to the clinician in the diagnosis of ILD. Regular CT scanners provide an image formed by the collection of 5 mm to 10 mm slices of tissue. High resolution CT is more defined with images formed from 0.75 mm to 1.5 mm slices. This provides a degree of detail required to recognize parenchymal patterns present in ILD.2 For example, the benefit of HRCT allows the clinician to visualize the abnormalities present in IPF, which include subpleural reticular opacities, traction bronchiectasis, and macrocystic honeycombing.11 The reliability of HRCT varies with the number of interobservers and the availability of other information, such as pathology and clinical information at the time the CT scan is read. This was apparent in one study that addressed a multidisciplinary approach to the diagnosis of ILD. As the number of participants (clinician, radiologist, and pathologist) increased, and the available data (HRCT, clinical information, and pathology) increased, interobserver variability in interpretation decreased.12 In other words, a panel approach increases the likelihood that a consensus would be reached on a diagnosis. Another study found that 80% of patients diagnosed with IPF based on HRCT were confirmed with a surgical lung biopsy.7 This degree of correlation between a noninvasive procedure such as HRCT and histological findings from a surgical biopsy has begun to reshape the method of diagnosis for some ILDs. It is clear that a struggle exists between diagnosing ILD based on HRCT alone versus open lung biopsy, both accompanied by clinical data. The ERS/ATS report does present a diagnostic pathway for the diagnosis of diffuse parenchymal lung diseases and includes those with known etiology and the IIPs.3
Pulmonologists have relied on the diagnostic potential of radiography, histopathology, and pulmonary function testing (PFT) for decades. From the chest radiograph to the CT scanner to the PFT laboratory and operating room, the struggle for the best method(s) to establish a definite diagnosis of ILD may be reaching a new frontier. Biomarkers are demonstrating possible aid to clinicians in not only the diagnosis of ILD but its progression and prognosis. One such biomarker, KL-6, is expressed on type 2 pneumocytes and bronchial epithelial cells.13 In ILDs, type 2 pneumocytes replace type 1 pneumocytes, and this may result in a higher KL-6 level in bronchial alveolar lavage and serum of ILD patients. This could further support the need for bronchoscopy in the diagnostic process, now seeking to collect biomarkers rather than cells and tissue samples. The study concluded, “KL-6 level may provide simple yet valuable information by which to identify patients with ILDs who are at risk for subsequent mortality.”13 During the study, 58 of 219 patients who died of respiratory failure had elevated levels of KL-6. Biomarkers present a new diagnostic technique, especially if they can be measured serologically and could aid in the diagnosis of and serial monitoring of ILDs.
The understanding of ILD is a process that continues to unfold in the areas of classification and diagnostics. Great efforts have been made by the ERS and the ATS to bring to the forefront a need for consensus on classification and diagnostic pathways. Striving for excellence in these areas for ILDs that are both aggressive and have a poor prognosis is a first step toward a more optimistic prognosis for patients.
Michael T. Provencher, BS, RRT, is staff respiratory therapist, Bryan LGH West Medical Center, Lincoln, Neb; and Paul F. Nuccio, MS, RRT, FAARC, is director of pulmonary services, Brigham and Women’s Hospital, Boston. For further information, contact [email protected].
References
- Nuccio P. Interstitial lung diseases. RT: For Decision Makers in Respiratory Care. 2007;20(1):36-8.
- Ryu J, Daniels C, Hartman T, Yi E. Diagnosis of interstitial lung diseases. Mayo Clin Proc. 2007;82:976-86.
- American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002;165:277-304.
- Churg A, Muller N. Cellular vs fibrosing interstitial pneumonias and prognosis. Chest. 2006;130:1566-70.
- Brown L, Schwarz M. Classifying interstitial lung diseases: remembrance of things past. Chest. 2006;130:1289-91
- Seong Y, Gee Y, Jae C, et al. Usefulness of open lung biopsy in mechanically ventilated patients with undiagnosed diffuse pulmonary infiltrates: influence of comorbidities and organ dysfunction. Crit Care. 2007;11:R93.
- Khalil N, O’Connor R. Idiopathic pulmonary fibrosis: current understanding of the pathogenesis and the status of treatment. CMAJ. 2004;171:153-60.
- Sigurdsson M, Isaksson H, Gudmundsson G, Gudbjartsson T. Diagnostic surgical lung biopsies for suspected interstitial lung disease: a retrospective study. Ann Thorac Surg. 2009;88:227-32.
- Park JH, Kim DK, Kim DS. Mortality and risk factors for surgical lung biopsy in patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg. 2007;31:1115-9.
- Noth I, Martinez F. Recent advances in idiopathic pulmonary fibrosis. Chest. 2007;132:637-50.
- Mueller-Mang C, Grosse C, Schmid K, Stiebellehner L, Bankier A. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics. 2007;27:595-615.
- Flaherty K, King T, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170:904-10.
- Satoh H, Kurishima K, Ishikawa H, Ohtsuka M. Increased levels of KL-6 and subsequent mortality in patients with interstitial lung disease. J Intern Med. 2006;260:429-34.








