Pulmonary fibrosis (PF) is a complex interstitial lung disease that afflicts an estimated 100,000 Americans, with 30-40,000 new cases diagnosed each year.
By Bill Pruitt, RRT, CPFT, AE-C, FAARC
Pulmonary fibrosis (PF) is a complicated and complex topic. PF and interstitial lung disease are often used interchangeably and may also be called diffuse parenchymal lung disease.[1-3] PF has many subtypes or disease entities (publications give a range from 150 to 200 different entities) but one in particular—idiopathic pulmonary fibrosis (IPF)—has been the subject of much of the published research as it accounts for 60% of the cases of PF.[1] If an underlying cause cannot be identified the term IPF is used.

With a diagnosis of pulmonary fibrosis, prognosis is poor. In cases of IPF, patients survive on average 2-4 years after diagnosis with a mean life expectancy of 3.8 years.[4-5] This article will take a close look at PF and IPF to examine the pathology, causes and risk factors, diagnosis, treatment, and future possibilities for patients with these diseases.
Anatomical and Pathological Changes
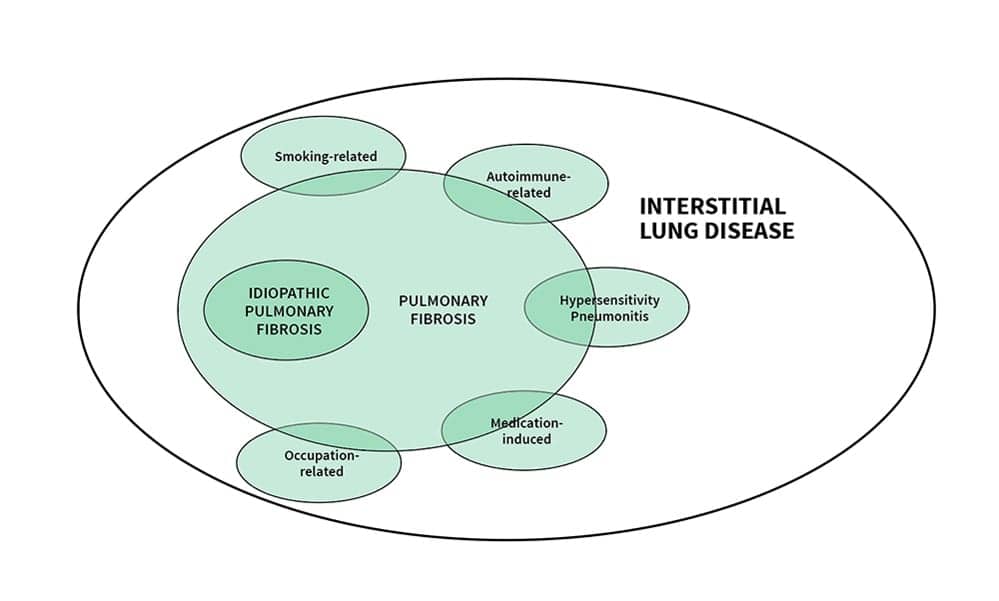
PF involves inflammation and progressive changes in the lung interstitium that brings about progressive and irreversible fibrotic changes. There are many overlapping characteristics of the subtypes of PF which often make it difficult to arrive at a clear diagnosis. (See Figure 1.)
Granulomas, honeycombing, and cavities are found in the lung parenchyma which are seen in high resolution computer tomography scans (HRCT). The interstitial space is thickened and the lung compliance is reduced as the fibrosis and inflammation worsens. In IPF there is a migration and proliferation of fibroblasts which differentiate into myofibroblasts.6
In PF diseases that involve inflammation as a major pathology, organizing pneumonia is seen.2 The decrease in compliance and fibrotic changes cause a decrease in all lung volumes when examined in the PFT lab. In addition, the diffusion capacity in the lungs is almost always reduced due to a thickened interstitial space along with ventilation/perfusion mismatching (from hypoxemia causing capillary shunting away from the affected areas).2-3 This decrease in diffusion capacity is seen in the DLCO test (however, there are two exceptions to this: when looking at Goodpasture’s syndrome and idiopathic pulmonary hemosiderosis, both of these diseases cause an increase in DLCO related to an increase in hemoglobin in the pulmonary tissues due to pulmonary hemorrhage).3
With the chronic hypoxemia and resulting pulmonary vasoconstriction, an increased load is put on the right heart which can cause right heart enlargement. The hypoxemia may also lead an increase in red blood cell (RBC) production (polycythemia). This combination of increased right heart enlargement and higher blood viscosity due to polycythemia can result in cor pulmonale for patients with PF.3
Causes and Risk Factors of Pulmonary Fibrosis
Many factors have been linked to PF including environmental exposures (including asbestos, coal dust, silica dust, moldy hay, wood pulp, and bird droppings) smoking, radiation, to connective tissue or autoimmune disorders (such as rheumatoid arthritis, scleroderma, and sarcoidosis), the use of certain medications (including chemotherapeutic drugs, cardiovascular drugs, and certain antibiotics) and to illicit drugs (such as heroin and methadone).2,7 Gastroesophageal reflux disease (GERD) may also play a role in the development of PF and IPF, and there may be a genetic cause in some cases.4,8,9 Many different charts, tables, and schematics have been produced to try and organize how to approach the causes and subtypes. An Internet search for images related to PF will reveal a variety of approaches used to try and clarify the complexities involved. Older, male patients of no particular race tend to have more risk of IPF with some two-thirds of the cases occurring in patients over 60 years old.9
Diagnosis
Diagnosing PF and IPF is challenging and many published articles on these issues agree that the best approach to making a clear diagnosis should involve a collaborative approach involving a pulmonologist, radiologist, and pathologist.5
The clinical presentation is non-specific and as a result symptoms may point to other possible issues. Patients have progressive dyspnea on exertion, a cough, and breath sounds reveal crackles. Some patients may develop clubbing in the fingertips. A thorough history with a close look at possible exposures is essential to determine if an external cause is involved.
Early in the disease, PFT results may be normal or near normal but as the disease progresses a complete PFT (spirometry, lung volumes, DLCO) will show a restrictive pattern and reduced lung volumes (decreased TLC, FRC, and RV seen in body plethysmography or in the nitrogen washout or helium dilution tests). In spirometry the FVC will be decreased while the FEV1 may be either normal or show a proportional decrease; so the FEV1/FVC may be normal or increased. The DLCO is also often reduced which reflects limited oxygenation so PaO2, SaO2, and SpO2 results are also low (two exceptions in the DLCO were mentioned earlier). The 6-minute walk test (6MWT) and cardiopulmonary exercise testing (CPET) are often used to assess a patient’s functional capacity and as disease progresses these test show increasing limitation for functional capacity.10
The collective PFT results have proven to be very helpful in making the diagnosis and in monitoring progression of PF over time. A decrease in FVC of 5% to 10% over 24 weeks is considered to be a significant change and relates to a twofold increase in mortality in the next year. A drop in DLCO or more than 15% also points to increased mortality.4,8 An initial assessment of pulmonary function and regular reassessment (possibly as often as every three months) is valuable in checking on the patient’s status.
High-resolution computer tomography scans (HRCT) show granulomas, honeycombing in the bases, and cavities in the lung parenchyma, along with volume loss. HRCT can also help distinguish between the different subtypes of PF and is considered the gold standard in assessing lung characteristics in PF and IPF.4,11 Bronchoscopy and bronchoalveolar lavage may help to rule in/rule out PF and also distinguish between various subtypes.11 Serologic tests can aid in distinguishing if the PF is due to connective tissue or autoimmune disorders.
Taking all of these measurements and tests into consideration, the diagnosis can be made for PF/IPF.
Treatment
There is currently no cure for PF or IPF. The clinical course for patients with IPF has been described in three scenarios.
Scenario One: the patient has a rapid decline in lung function with the time from onset of the disease (with a short period of being asymptomatic), to onset of symptoms and diagnosis, to death occurring in a year or less.
Scenario Two: the patient has the onset of disease but has a slow decline in lung function and may be asymptomatic until an exacerbation hits. At this turn, symptoms become very noticeable and the diagnosis is made. Again, there is a period of slow decline but another exacerbation causes a profound increase in symptoms and loss of lung function. Often, this ends in death and can occur in one to three years after onset.
Scenario Three: the overall course of the disease is a slow progression with steady decline in lung function and more gradual increase in symptoms until eventually death occurs. This may occur over the course of four to six years.12
Pirfenidone and nintedanib received USA FDA approval in 2014 as treatment agents and are currently the two best options for addressing PF/IPF. Both are antifibrotics and have shown an impact on reducing the rate of decline in FVC.13,14 Pirfenidone is given orally and has shown a reduction in the decline in the 6MWT distance improved the time for progression-free survival (defined as death or decrease in vital capacity of > 10%). Nintedanib is also given orally and beyond reducing the rate of decline in the FVC, this agent also reduced the number of acute exacerbations.14
Previously, treatment for IPF included administration of azathioprine (immunosuppressive agent), prednisone (corticosteroid, anti-inflammatory agent), and N-acetylcysteine (mucolytic). However, in a study published in 2012, researchers found that this approach resulted in an increased risk of death and hospitalization compared to placebo; therefore the three-drug combination is no longer recommended for IPF.13,14 Supplemental oxygen is indicated for those who have hypoxemia and high-flow nasal cannula therapy or noninvasive ventilation may be useful in treating acute exacerbations that involve refractory hypoxemia.16
Comorbid conditions also need consideration in the plan of care to improve overall health of the patient (particularly any cardiopulmonary disorder and/or GE reflux issues). When the disease subtype is defined in PF, the treatment plan is established to target the specific disease. In those patients with a known cause it is important to remove the exposure if possible, and for all who smoke, successful smoking cessation is needed.
Pulmonary rehabilitation has been shown to increase exercise tolerance and improve symptoms and quality of life for those with IPF, as seen in a recent systematic review and meta-analysis published in 2018.17 For patients with end-stage fibrosis, lung transplant is considered a last resource option.
Future Possibilities
Many agents are being investigated to see if any can improve the prospects of patients with PF and IPF. Inhaled interferon, carbon monoxide, Angiotensin-converting enzyme (ACE) inhibitors and receptor blockers (ARB) agents, anticoagulants, thalidomide, tumor necrosis factor inhibitors, and sildenafil are among the pharmacologic choices in various research projects.
As more is known about the molecular changes that occur in the lung tissues, better options may be uncovered. As seen with the work in inhaled interferon, respiratory therapists may be involved in research and administration of some of these new targeted therapies as inhaled agents.18
Conclusion
PF and IPF are rare diseases but they have dire consequences for those diagnosed with these problems. The specific diagnosis is important if the subtype can be identified. Comorbid conditions need to be addressed as well to improve the quality of life and state of health. Pharmacologic agents, oxygen therapy, pulmonary rehabilitation, and smoking cessation can help in these cases but considering the often rapid decline and death of these patients it is prudent to discuss palliative and hospice care.
Familiarity with the symptoms and PFT data for PF and IPF are important for respiratory therapists since we often work with these patients in providing care and support.
RT
Bill Pruitt, RRT, CPFT, AE-C, FAARC is a senior instructor and director of clinical education in the department of Cardiorespiratory Sciences, College of Allied Health Sciences, at the University of South Alabama in Mobile. He is also currently an elected member of the Board of Directors for the National Asthma Educator Certification Board (NAECB). For more information, contact [email protected].
References
- 1. Danoff SK, Terry PB, Horton MR. A clinician’s guide to the diagnosis and treatment of interstitial lung diseases. Southern Medical Journal. 2007 Jun 1;100(6):579-88.
- 2. Karpel, S., Linz, A. (2020) Interstitial Lung Disease. Linz’s comprehensive Respiratory Diseases. Jones & Bartlett Learning. Burlington MA.
- 3. Des Jardins, T., & Burton, G. (2016) Interstitial Lung Diseases. Clinical Manifestations and Assessment of Respiratory Disease, 7th ed. Elsevier. St. Louis MO.
- 4. Meyer KC. Diagnosis and management of interstitial lung disease. Translational respiratory medicine. 2014 Dec 1;2(1):4.
- 5. Salvatore M, Ishikawa G, Padilla M. Is It Idiopathic Pulmonary Fibrosis or Not?. The Journal of the American Board of Family Medicine. 2018 Jan 1;31(1):151-62.
- 6. Hospenthal MA. Diagnosis and management of idiopathic pulmonary fibrosis: Implications for respiratory care. Respiratory care. 2006 Apr 1;51(4):382-91.
- 7. Mottram, C. (2018) Indications for Pulmonary Function Testing. Ruppel’s Manual of Pulmonary Function Testing, 11th ed. Elsevier. St. Louis MO.
- 8. Vega-Olivo M, Criner GJ. Idiopathic pulmonary fibrosis: A guide for nurse practitioners. The Nurse Practitioner. 2018 May 17;43(5):48.
- 9. Hospenthal MA. Diagnosis and management of idiopathic pulmonary fibrosis: Implications for respiratory care. Respiratory care. 2006 Apr 1;51(4):382-91.
- 10. Vainshelboim B, Myers J, Oliveira J, Izhakian S, Unterman A, Kramer MR. Physiological Responses and Prognostic Value of Common Exercise Testing Modalities in Idiopathic Pulmonary Fibrosis. Journal of cardiopulmonary rehabilitation and prevention. 2019 May 1;39(3):193-8.
- 11. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. American journal of respiratory and critical care medicine. 2018 Sep 1;198(5):e44-68.
- 12. Ley B, Collard HR, King Jr TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2011 Feb 15;183(4):431-40.
- 13. Morisset J, Lee JS. New trajectories in the treatment of interstitial lung disease: treat the disease or treat the underlying pattern?. Current opinion in pulmonary medicine. 2019 Sep;25(5):442-9.
- 14. King CS, Nathan SD. Practical considerations in the pharmacologic treatment of idiopathic pulmonary fibrosis. Current opinion in pulmonary medicine. 2015 Sep 1;21(5):479-89.
- 15. Raghu G. Idiopathic pulmonary fibrosis: new evidence and an improved standard of care in 2012. The Lancet. 2012 Aug 18;380(9842):699-701.
- 16. Imai R, Tsugitomi R, Nakaoka H, Jinta T, Tamura T. Noninvasive Oxygenation Strategies For Acute Exacerbation of Interstitial Lung Disease: A Retrospective Single-center Study and a Review of the Literature. Clinical Pulmonary Medicine. 2019 May 1;26(3):87-91.
- 17. Gomes-Neto M, Silva CM, Ezequiel D, Conceição CS, Saquetto M, Machado AS. Impact of pulmonary rehabilitation on exercise tolerance and quality of life in patients with idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Journal of cardiopulmonary rehabilitation and prevention. 2018 Sep 1;38(5):273-8.
- 18. Staitieh BS, Renzoni EA, Veeraraghavan S. Pharmacologic therapies for idiopathic pulmonary fibrosis, past and future. Annals of medicine. 2015 Feb 17;47(2):100-5.