Asthma patients show variable responses to all asthma therapies partly because of real biological variation in the underlying mechanisms of their disease.
 The randomized, double-blind, placebo-controlled clinical trial is the keystone of drug development to determine clinical efficacy, and increasing interest is being paid to effectiveness studies that take into account real-world factors (including patient adherence). Both the efficacy and the effectiveness of drug therapy vary among patients, with subgroups of patients showing either incomplete or consistently above-average responses. Such intersubject variability may be attributed to diagnostic error, errors in assessing response, and differences in patient adherence, but a substantial component is likely to result from genuine biological variation in disease mechanisms, some of which may be genetic in origin. In addition, adverse effects of drug therapy are not uncommon and may also have a genetic basis.1 Characterization of genetic mechanisms may allow screening to provide better targeting of drugs toward those most likely to benefit, and away from those most likely to experience serious adverse effects. An example from the asthma field is the glycine (Gly)/arginine (Arg)-16 genetic polymorphism in the beta-2-adrenoceptor, such that patients with Arg-16 polymorphism show enhanced responses to inhaled beta2-agonist therapy, probably because of differences in downregulation of the receptor in airway smooth-muscle cells.2
The randomized, double-blind, placebo-controlled clinical trial is the keystone of drug development to determine clinical efficacy, and increasing interest is being paid to effectiveness studies that take into account real-world factors (including patient adherence). Both the efficacy and the effectiveness of drug therapy vary among patients, with subgroups of patients showing either incomplete or consistently above-average responses. Such intersubject variability may be attributed to diagnostic error, errors in assessing response, and differences in patient adherence, but a substantial component is likely to result from genuine biological variation in disease mechanisms, some of which may be genetic in origin. In addition, adverse effects of drug therapy are not uncommon and may also have a genetic basis.1 Characterization of genetic mechanisms may allow screening to provide better targeting of drugs toward those most likely to benefit, and away from those most likely to experience serious adverse effects. An example from the asthma field is the glycine (Gly)/arginine (Arg)-16 genetic polymorphism in the beta-2-adrenoceptor, such that patients with Arg-16 polymorphism show enhanced responses to inhaled beta2-agonist therapy, probably because of differences in downregulation of the receptor in airway smooth-muscle cells.2
As has been the case for other asthma therapies, including both beta2-agonists and glucocorticoids, clinical trials of leukotriene-receptor antagonists (LTRA) demonstrate a range of patient responses to these drugs. Genetic variations in enzymes of the leukotriene synthetic pathway may cause some individuals with asthma to produce cysteinyl-leukotrienes (cys-LTs) to a greater extent than other patients, predisposing them to more severe asthma symptoms (particularly poorer lung function), and perhaps defining those individuals who may show the best clinical responses to LTRA therapy.
Leukotrienes as Asthma Mediators
The cys-LTs LTC4 and LTD4 are the most potent bronchoconstrictor mediators ever described, being two to three orders of magnitude more potent than histamine, prostanoids, or platelet-activating factor in human airways. They also have a wide range of proinflammatory actions relevant to asthma and allergic disease, including mucus hypersecretion, impaired mucociliary clearance, vascular hyperpermeability, neuropeptide release, eosinophil recruitment, bronchial smooth-muscle hypertrophy, and fibroblast collagen deposition.3 Cys-LTs are synthesized via the 5-lipoxygenase (5-LO) pathway (Figure 1, page 31). Cell activation leads to the liberation of arachidonic acid from membrane phospholipids, and 5-LO converts arachidonate to LTA4, with 5-LO activating protein as a cofactor. In mast cells, eosinophils, basophils, and monocyte-macrophages, LTA4 is conjugated to glutathione to form the first of the cys-LTs, LTC4, by LTC4 synthase.4 Extracellular conversion of LTC4 to LTD4 and then to LTE4 produces a family of cysteine-containing LTs (formerly known as slow-reacting substance) that act at specific cys-LT1 receptors on target tissues, such as bronchial smooth muscle.5
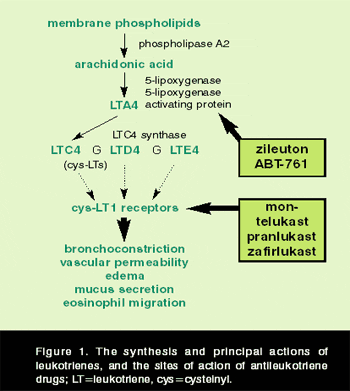
Leukotriene-modifier drugs include the 5-LO inhibitor zileuton and the LTRAs montelukast, pranlukast, and zafirlukast. In bronchoprovocation models, these drugs significantly block bronchoconstrictor responses to inhaled allergens, sulfur dioxide, nonsteroidal anti-inflammatory drugs (NSAIDs), platelet-activating factor, and exercise/cold air in susceptible individuals.6 Their efficacy in multiple-dose trials in clinical asthma has been extensively reviewed,7 with improvements of 5% to 15% typically noted in baseline lung function and improvements of 20% to 60% seen for secondary outcomes measures, including beta-agonist use, exacerbations, daytime symptom scores, night awakenings, and absences from work and school. Anti-inflammatory activity is reflected by reductions in eosinophil counts in blood, induced sputum, and bronchial biopsy samples of patients treated with LTRAs.
Clinical responses to antileukotriene drugs vary from subject to subject, even when these patients are ostensibly of similar clinical phenotype and severity. In a direct comparative trial of inhaled beclomethasone (400 to 500 µg per day) and oral montelukast (10 mg per day) in mild-to-moderate adult asthma for 12 weeks, basal values for forced expiratory volume in 1 second (FEV1) improved an average of 13% from pretrial values in the corticosteroid group and about 7% in the montelukast group.8 Superficially, it might be concluded that montelukast has lower efficacy in these patients, but examination of the distribution of responses shows that the higher average improvement in the steroid-treated group was entirely driven by a very small minority of patients (10%) who showed extremely large improvements (>40%) in FEV1. Over most of the range of responses, beclomethasone and montelukast responses were very similar. FEV1 deteriorated in a significant proportion of patients treated with either drug, while montelukast was marginally more effective than beclomethasone in patients who showed improvements in FEV1 of 10% to 40%. Understanding the mechanisms of such variation will allow therapy to be individualized for each patient to maximize the probability of a beneficial response. In contrast, relying on averaged data from clinical trials may lead to suboptimal drug choices for a substantial proportion of patients. In other words, relatively few patients show an average response to therapy, but the tyranny of the mean can eventually lead to treatment guidelines and health care systems that place restrictions on clinical freedom to choose the best drug for an individual patient. Genetic testing may provide the rationale for better targeting of drugs to individual patients and thus broaden clinical choice.
Genetics of 5-LO
Genetic factors are likely to underlie much of the variable response to asthma medications. Polymorphism (a mutation with a prevalence greater than 1% in a given population) may occur in gene-coding regions, and such polymorphisms may alter the amino-acid sequence of the protein, leading to abnormal enzyme or receptor function. An example is Arg/Gly-16 beta2 receptor polymorphism previously noted.2 Polymorphism may also occur in the regulatory region of a gene, often situated upstream from the transcription-initiation site; here, the variant forms of the gene may lead to altered transcription of the gene such that the final protein is expressed in abnormal amounts (compared with the amounts produced by individuals carrying the more common, or wild-type, allele). In 1996, a polymorphism in the 5-LO gene promoter was identified, consisting of a variable number of tandem repeats of GC-rich motifs necessary for the activity of the Sp1 and Egr1 transcription factors.9 The wild-type allele had five such tandem motifs, while variant alleles with only three or four motifs—or with six motifs—had lower transcriptional activity in gene-reporter assays. In 681 asthma patients, the wild-type allele had a prevalence of 81.7%, with the alleles of three, four, and six motifs having prevalence rates of 14.7%, 3.3%, and 0.3% respectively. The prevalence of the wild-type and variant alleles did not differ among normal subjects, asthma patients, and aspirin-intolerant asthma (AIA) patients, suggesting that the variant alleles do not predispose subjects to an asthma phenotype.9 Nevertheless, in a clinical trial10 of the oral 5-LO inhibitor ABT-761 for 12 weeks in 681 asthma patients, patients who had one or two copies of the wild-type allele showed a significant mean improvement in FEV1 (approximately 15%), compared with placebo (5%), while patients who had two copies of the variant alleles showed no change in FEV1. Thus, the variant alleles are recessive, and homozygosity for the variant alleles confers a relative lack of response to anti-leukotriene therapy in approximately 5% to 6% of subjects with asthma. This small subgroup of patients must have a form of asthma in which airway narrowing is produced by mediators other than leukotrienes.
LTC4 synthase is the downstream enzyme that synthesizes the first of the cys-LTs, LTC4. Very high expression of LTC4 synthase has been described in the airway walls of patients with AIA.11 These patients may experience life-threatening asthma attacks after ingesting aspirin or other NSAIDs, but they also tend to have moderate-to-severe chronic asthma even when NSAIDs are avoided.12 The acute episodes may be triggered by inhibition of the synthesis of a protective prostaglandin,13 but both the acute attacks and the chronic disease are associated with high levels of cys-LT production.12
A promoter polymorphism in the LTC4 synthase gene has been identified.14 It consists of a substitution of the base cytosine for adenosine at a position 444 bases upstream from the transcription-initiation site. The polymorphism is known as -444A/C, with A being the wild-type and C the variant allele. Patients with AIA are significantly more likely to have one or two of the variant C alleles (75%) than asthma patients who are not aspirin-sensitive (44%) or normal subjects (42%). The variant LTC4 synthase allele thus represents a significant risk factor for aspirin intolerance (odds ratio 3.89, P<.05). These prevalence data15 from a Polish population have not, however, been confirmed by other investigators15 in patients from Japanese populations or US white populations. Thus, whether LTC4 synthase predisposes individuals to the aspirin-asthma phenotype remains open to doubt.
Based on the observation that the LTC4 synthase C-444 allele is also represented in 42% to 44% of aspirin-tolerant asthma patients and normal subjects, however, we hypothesized that it may not predispose subjects to aspirin intolerance directly, but rather to cys-LT overproduction in any asthma phenotype when exposed to the relevant trigger. To verify this hypothesis, the polymorphism must be demonstrated to act at the levels of messenger RNA (mRNA), protein, cellular LT synthesis, lung function in vivo, and clinical response to antileukotriene therapy. When transfected into HeLa cells, the C-444 allele significantly increases transcription of a luciferase reporter gene, compared with the A-444 allele.16 In AIA patients, mRNA for LTC4 synthase is overexpressed in blood eosinophils from patients with C-444 allele(s), compared with those from patients with the wild-type genotype (A/A).17 This is associated with higher urinary LTE4 levels. In normal human eosinophils in vitro, LTC4 synthesis following calcium ionophore stimulation in the presence of indomethacin is three times higher in cells from subjects with one or two variant LTC4 synthase alleles (A/C or C/C) than it is in cells from subjects with wild-type alleles (A/A).18 Furthermore, in 317 mild-to-moderate asthma patients, FEV1 was significantly lower in patients with the variant LTC4 synthase genotypes (92.7% of the predicted values, n=162, P=.004) than in patients with the wild-type genotype (97.5% of the predicted values, n=155).16 Thus, the LTC4 synthase polymorphism may predict baseline lung function in asthma patients and point to a subgroup of asthma patients (with a prevalence of around 50%) in whom cys-LTs play a particularly important pathophysiological role.
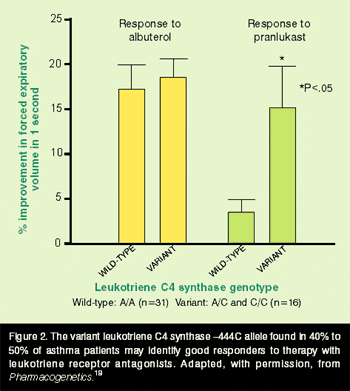
To investigate this further, we conducted a small open study of23 patients with chronic severe asthma whose lung-function responses to 2 weeks of treatment with oral zafirlukast (20 mg, given twice daily) were compared with prestudy baseline values. In those patients with variant LTC4 synthase genotypes (n=13), post-zafirlukast improvements in FEV1 (+9%) and forced vital capacity (+15%) tended to be greater than in the 10 patients with wild-type genotypes (who had decreases of 12% and 18%, respectively).18 Although these differences were not significant in this small study, the data do suggest that the LTC4 synthase genotype can predict lung-function responses to antileukotriene therapy. The outcome has been confirmed in a larger study,19 which reported the responses to 4 weeks’ treatment with oral pranlukast in 48 patients with mild-to-moderate asthma. Mean FEV1 improved only 2.8% in wild-type patients, but those with the variant LTC4 synthase genotypes showed an improvement of 12.4% (P=.05), with half of them increasing their FEV1 by at least 10% (P=.03), as shown in Figure 2. Polymorphism in LTC4 synthase was a highly significant predictor of lung-function improvement after pranlukast treatment. The large subgroup of patients who have the variant genotype may represent a phenotype of leukotriene-driven asthma and, hence, define the optimal target population for LTRA therapy. Further testing of this hypothesis is needed in larger groups of patients, and should include a wider range of asthma measures than FEV1 alone.
Summary
Asthma patients show variable responses to all asthma therapies partly because of real biological variation in the underlying mechanisms of their disease. Intersubject variability in response to antileukotriene therapy is comparable to that seen for glucocorticoids and beta2-agonists, and may be accounted for, at least in part, by promoter polymorphisms in the 5-LO and LTC4 synthase genes. The good safety profile of LTRAs provides no obstacle to physicians’ assessing an oral LTRA in appropriate patients over a period of weeks or months to judge its effectiveness in the real-world setting. In the future, although there are formidable ethical and other hurdles to surmount, such assessments may also include genetic testing to help predict the probable response to LTRAs.
Anthony P. Sampson, PhD, is the director of Allergy and Inflammation Research in the Division of Infection, Inflammation & Repair, University of Southampton School of Medicine, Southampton, England.
References
1. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200-1205.
2. Martinez FD, Graves PE, Baldini M, Solomon S, Erickson R. Association between genetic polymorphisms of the beta2-adrenoceptor and response to albuterol in children with and without a history of wheezing. J Clin Invest. 1997;100:3184-3188.
3. Hay DWP, Torphy TJ, Undem BJ. Cysteinyl leukotrienes in asthma: old mediators up to new tricks. Trends Pharmacol Sci. 1995;16:304-309.
4. Penrose JF, Spector J, Baldasaro M, et al. Molecular cloning of the gene for human leukotriene C4 synthase: organization, nucleotide sequence, and chromosomal localization to 5q35. J Biol Chem. 1996;271:11356-11361.
5. Lynch KR, O’Neill GP, Liu Q. et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789-793.
6. Holgate ST, Bradding P, Sampson AP. Leukotriene antagonists and synthesis inhibitors: new directions in asthma therapy. J Allergy Clin Immunol. 1996;98:1-13.
7. Drazen JM, Israel E, O’Byrne PM. Treatment of asthma with drugs modifying the leukotriene pathway. N Engl J Med. 1999;340:197-206.
8. Malmstrom K, Rodriguez-Gomez G, Guerra J, et al. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma: a randomized, controlled trial. Ann Intern Med. 1999;130:487-495.
9. In K, Asano K, Beler D, et al. Naturally-occurring mutations in the human 5-lipoxygenase gene promoter that modify transcription factor binding and reporter gene transcription. J Clin Invest. 1997;99:1130-1137.
10. Drazen JM, Yandava CN, Dube L, et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat Genet. 1999;22:168-170.
11. Cowburn A, Sladek K, Soja J, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101:834-846.
12. Szczeklik A, Stevenson DD. Aspirin-induced asthma: advances in pathogenesis and management. J Allergy Clin Immunol. 1999;104:5-13.
13. Sestini P, Armetti L, Gambaro G, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153:572-575.
14. Sanak M, Simon HU, Szczeklik A. Leukotriene C4 synthase promoter polymorphism and risk of aspirin-induced asthma. Lancet. 1997;350:1599-1600.
15. Yoshida S, Penrose JF, Stevenson DD, et al. Polymorphism with genes in cysteinyl leukotriene synthesis pathway in aspirin-intolerant asthma. Am J Respir Crit Care Med. 2000;161:A602.
16. Sayers I, Hayward B, Van Eerdewegh P, et al. Allelic association and functional studies of promoter polymorphism in the leukotriene C4 synthase gene (LTC4S) in asthma. Thorax. In press.
17. Sanak M, Pierzchalska M, Bazan-Socha S, Szczeklik A. Enhanced expression of the leukotriene C4 synthase due to overactive transcription of an allelic variant associated with aspirin-intolerant asthma. Am J Respir Cell Mol Biol. 2000;23:290-296.
18. Sampson AP, Siddiqui S, Buchanan D, et al. Variant LTC4 synthase allele modifies cysteinyl-leukotriene synthesis in eosinophils and predicts clinical response to zafirlukast. Thorax. 2000;55:S28-S31.
19. Asano K, Shiomi T, Hasegawa N, et al. Leukotriene C4 synthase gene A-444C polymorphism and clinical response to a CYS-LT1 antagonist, pranlukast, in Japanese patients with moderate asthma. Pharmacogenetics. 2002;12:565-570.