|
For many years, it seemed that every article or textbook chapter on cystic fibrosis started out with the tired old saw, “Cystic Fibrosis is the most common fatal inherited disease of Caucasians.” It is indeed common, with a gene frequency among North American Caucasians of approximately one in 31 individuals. It is estimated that 30,000 individuals in the United States have the disease.1 It is, at present, incurable, with no specific curative therapy available. Cystic fibrosis (CF) is widely viewed as profoundly life shortening, even though life expectancy has lengthened remarkably over the past several decades since the institution of a systematized plan of care. At present, the Cystic Fibrosis Foundation (CFF)1 reports the “mean predicted survival age” of an individual with CF in 2008 to be 37.4 years. This term is based on CFF registry data, and is defined as the age to which half the current population of patients with CF would be expected to survive.
The rise in predicted survival age has been remarkable, yet it has not been based on mutation-specific therapies that address the molecular defect in the protein that leads to the disease. The affected protein, named CFTR for “cystic fibrosis transmembrane conductance regulator,” was discovered in 1989. The recent 2009 North American Cystic Fibrosis Conference marked the 20th anniversary of this discovery with a mixture of optimism, excitement, and regret. Initial enthusiasm in 1989 led to a belief that a cure was “just around the corner.” Twenty years later, most clinicians who treat patients with CF never use the word cure in discussing the disease. We believe that improved treatments, especially the increased emphasis on airway clearance as a cornerstone of CF care, have led to improved outcomes. Yet treatments that directly address the molecular/cellular defect that leads to the dehydration of the periciliary fluid layer remain elusive. Hypertonic saline (nebulized 7% saline) appears to briefly reverse the desiccation of the periciliary fluid layer and improve mucociliary clearance. This therapy, which has gained widespread acceptance, does not directly affect CFTR but can be said to be the first FDA-approved therapy in this disease to directly address the cellular defect in CF.2,3
The CFF, in partnership with a number of small pharmacologic firms, has been funding preclinical and early clinical trials of several small molecule therapies that directly affect CFTR function. Because of the relatively small number of patients affected by CF, there has been little incentive for the large pharmaceutical companies to invest in CF medications. The concept of “venture philanthropy” has been championed by the CFF, and has led to several new potential therapies. Since 1998, the CFF has invested more than $300 million in drug research and has created an organization called the Therapeutics Development Network (TDN) to coordinate clinical research in the CF care centers. Together, the CFF and the TDN have established the new standard in research in rare diseases, and have one of the most successful programs in the world for clinical research and development of new therapies.
A summary of all therapies in development from the Cystic Fibrosis Foundation can be found in Figure 1. Preliminary data from these trials has led to a great deal of excitement in the field, despite the likelihood that at least some of these therapies will not apply to the majority of American patients. I will review some of the most promising of these new therapies in the CF “pipeline.”
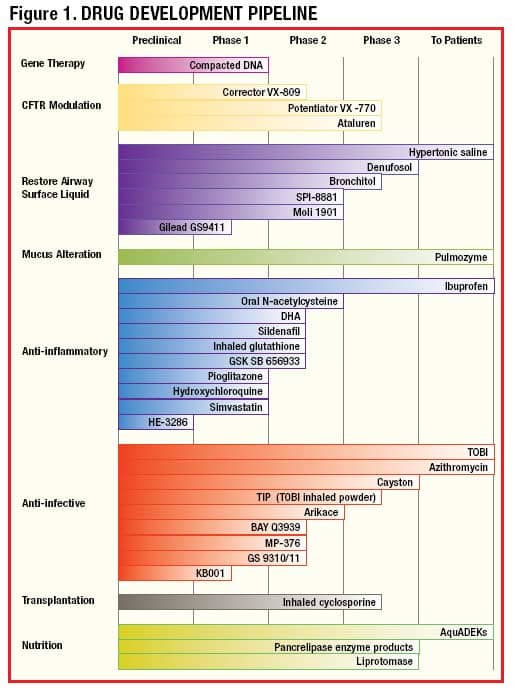 |
| Source: Cystic Fibrosis Foundation. Reprinted with permission. |
PTC-124/Ataluren
The small pharmaceutical firm PTC Therapeutics has been focused on small molecules that target post-transcriptional control processes (hence the name). Their first drug, initially called PTC-124 and later named Ataluren, promotes read-through in nonsense mutations—point mutations that lead to the formation of a premature stop codon in the message RNA of proteins, and tend to lead to truncated and dysfunctional proteins.
Nonsense mutations are the cause of CF in just 2% to 5% of cases in the United States, but are the most common causes of CF in Israel. Ataluren was found with a high throughput screening technique using a large library of small molecules. It is an orally administered medication. This molecule binds to the ribosome and allows read-through of premature stop codons while allowing for the function of normal stop codons. This drug has been studied in two diseases, cystic fibrosis and Duchenne/Becker muscular dystrophy, with studies planned in hemophilia A caused by nonsense mutations. The phase 2a trial in cystic fibrosis demonstrated a statistically significant improvement in CFTR function in the airways of subjects receiving the medication, with reduction in cough frequency and improvement in pulmonary function in a short-term administration of this medication.4
A 28-day “proof of concept” trial of this orally administered medication looked at adults with CF and at least one nonsense mutation in the CFTR gene. Chloride transport was evaluated by the electrophysiological technique, nasal potential difference. The investigators found that chloride transport entered the normal range in 13 of 23 subjects during the first treatment phase (dosed at 16 mg/kg/day for 14 days) and 9 of 21 in the second phase (dosed at 40 mg/kg/day for 14 days). This study was not powered for pulmonary function as an outcome.
PTC Therapeutics is currently enrolling nonsense mutation cystic fibrosis patients into a 48-week, randomized, double-blind, multicenter phase 3 trial of Ataluren.
VX-770 and VX-809
Some of the most dramatic results in recent years have been with the CFTR “potentiator” medication, VX-770.5 This orally available small molecule was created by Vertex Corp and was also discovered using high throughput screening. It targets deactivating mutations of CFTR in which the protein is expressed at the cell surface but does not function as a chloride channel. The prototypic mutation that responds to VX-770 is G551D, which occurs in 3% to 4% of patients with CF. A 14-day, phase 2 trial of VX-770 has been completed in a group of 20 patients with CF.
Significant improvements in lung function, sweat chloride, and nasal potential difference were found. A 10% improvement in lung function was seen in this small group of subjects, which is a remarkable finding given the short duration of the study. VX-770 is the first therapy in CF that has led to correction of sweat chloride results, an equally exciting result. A phase 3 trial of VX-770 has been initiated.
Vertex Corp is also studying a compound, VX-809, designed to increase the amount of CFTR that reaches the cell surface in patients with the Delta-F 508 mutation. Delta-F 508 is the most common mutation in North American patients with CF, occurring in more than 70% of affected individuals. This drug is currently being studied in a phase 2 trial. It is possible that a combination of VX-770 and VX-809 will improve CFTR function in patients with the most common mutation causing CF, but there are no data yet available for analysis.
Denufosol
Mutation of CFTR, which functions as a chloride channel, leads to decreased chloride secretion and a resulting increase in absorption of sodium though the epithelial sodium channel. The net result is a loss of sodium and chloride from the fluid that lines the airway epithelial cells, and with loss of salt there follows a loss of water. The level of airway surface liquid becomes too low for the cilia to effectively beat.
Denufosol tetrasodium is a novel chloride channel activator targeting CF patients of all CFTR genotypes that was discovered by Inspire Pharmaceuticals. It is a P2Y2 receptor agonist, which activates non-CFTR chloride channels in the airway, inhibits sodium absorption, and increases ciliary beat frequency. Activation of the P2Y2 receptor on airway epithelial cells leads to hydration of the extracellular fluid layer, hydration of mucous secretion, and increased mucociliary clearance. It is delivered by jet nebulization and administered three times daily. Phase 2 results were published in 2007.
In June 2008, the results of the TIGER-1 phase 3 trial were released. The trial was named TIGER for “Transport of Ions to Generate Epithelial Rehydration.” This name is well chosen, as it addresses the main disorder in CF: the desiccation of airway epithelial surface liquid. The TIGER-1 trial demonstrated a significant improvement in lung function in a 24-week treatment trial, with an additional 24-week open label add-on arm, involving 352 subjects with mild lung disease in 62 care centers. The treatment effect was modest but significant (an increase of just 45 ml in FEV1) in subjects receiving denufosol, and this improvement in FEV1 occurred without apparent abatement in patients who continued to receive this medication in the open label extension (see Figure 2). A second phase 3 study is fully enrolled and is ongoing. Denufosol has been granted “fast track” and “orphan drug” status, which should accelerate the approval process.
 |
| Figure 2. Mean Change from Baseline FEV1. Source: Inspire Pharmaceuticals. Reprinted with permission. |
Conclusion
The CFF’s venture philanthropy approach and organized therapeutic research through its TDN have led to some of the most exciting therapies that have been seen in this disease and, for some, a potential for cure. Although phase 3 trials are still in the works, hope is very high that this disease will someday be described as “previously incurable.” This remains a very exciting time for individuals caring for patients with cystic fibrosis, and a time of great optimism for families and patients with this disease.
Jonathan Finder, MD, is professor of pediatrics, University of Pittsburgh. For further information, contact [email protected].
References
- About Cystic Fibrosis. Cystic Fibrosis Foundation. Available at: www.cff.org. Accessed December 31, 2009.
- Dondalson S, Bennett W, Zeman K, Knowles M, Tarran R, Boucher R. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241-50.
- Flume PA, O’Sullivan BP, Robinson KA, et al. Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176:957-69.
- Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719-27.]
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825-30.
- Deterding RR, Lavange LM, Engels JM, et al; for the Cystic Fibrosis Therapeutics Development Network and the Inspire 08-103 Working Group. Phase 2 randomized safety and efficacy trial of nebulized denufosol tetrasodium in cystic fibrosis. Am J Respir Crit Care Med. 2007;176:362-4.